将过量的二氧化碳(CO2)转化为具有高附加值的化学原料是解决全球能源危机、实现碳中和的最佳方案之一。尽管自然界可以通过光合作用将CO2高效地转化为有机物,但是由于CO2具有热力学稳定性和化学惰性,人工捕获和还原CO2仍然是一项非常具有挑战性的工作。近期,永利欢乐娱人城崔刚龙教授团队采用了高精度的多参考CASPT2方法探索了铱(Ir)配合物催化剂[Ir(III)H]+的激发态性质和光物理过程,采用高效的DFT方法系统地研究了其后续生成HCOOH和CO的整个催化反应路径,同时还计算了辐射、无辐射和电子转移过程的相应速率,最终系统研究了Mes-IrPCY2光催化二氧化碳还原的微观反应机理,揭示了HCOOH高选择性的物理起源。
图1. 铱配合物[Ir-H]+光催化CO2还原。图片来源:Angew. Chem. Int. Ed.
[Ir(III)H]+初期的光物理过程可以用四态模型解释(S0,1MLCT,3LE和3MLCT)。光激发后,体系首先弛豫到Franck-Condon区附近的1MLCT极小点,随后通过两条无辐射通道(直接的1MLCT→3MLCT通道和3LE介导的1MLCT→3LE→3MLCT通道)到达能量最低的3MLCT态,即后续光催化反应的前驱态。随后,[Ir(III)H]+在3MLCT态可以与电子牺牲剂BIH发生单电子转移(SET)反应生成活性中间体OERS和BIH•+,OERS在BIH•+协助下进一步与CO2发生还原反应生成HCOOH。除SET反应外,[Ir(III)H]+在光激发到达3MLCT态后也可与溶剂分子发生脱质子反应,随后通过系间窜跃过程生成Ir(I)中间体,Ir(I)中间体在BIH•+协助与CO2反应生成CO。在光催化循环中,无辐射跃迁扮演着极其重要的角色。此外,计算表明HCOOH高选择性应来源于两个方面:HCOOH活性中间体OERS的有效反应和HCOOH生成路径的速控步能垒更低。计算发现SET过程生成的BIH•+通过提供质子或电子在关键的还原反应中显著降低了能垒。本研究表明,由于反应涉及电子激发态、多自旋态、开壳层中间体和显著的非绝热效应,高精度的多参考电子结构方法对于研究有机金属化合物光催化反应的部分阶段是不可或缺的。最后,所获得的机理有助于化学家进一步理解、调控和设计基于功能集成分子催化剂的光催化二氧化碳还原反应。
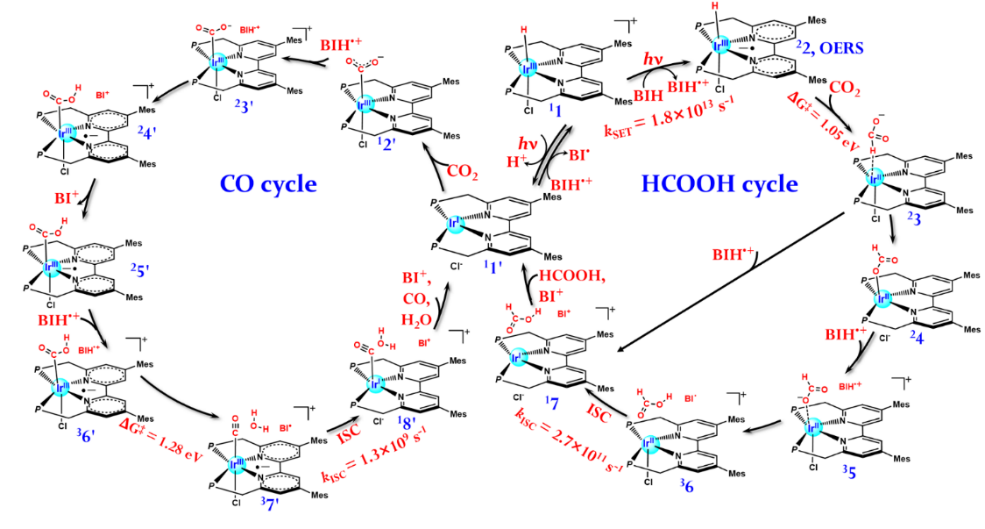
图2. 计算推测的光催化CO2还原生成甲酸和一氧化碳的反应机理。图片来源:Angew. Chem. Int. Ed.
这一研究成果近期发表在Angewandte Chemie International Edition上,文章第一单位是永利欢乐娱人城永利欢乐娱人城,通讯作者是崔刚龙教授,第一作者是其博士生彭灵雅同学。工作得到了科技部、基金委、永利欢乐娱人城的经费资助。
论文信息:https://doi.org/10.1002/anie.202315300.
Photocatalytic Reduction of CO2 to HCOOH and CO by a Phosphine-Bipyridine-Phosphine Ir(III) Catalyst: Photophysics, Nonadiabatic Effects, Mechanism, and Selectivity. Ling-Ya Peng, Guang-Ning Pan, Wen-Kai Chen, Xiang-Yang Liu, Wei-Hai Fang, and Ganglong Cui, Angew. Chem. Int. Ed. 2023, e202315300.